









Efnisyfirlit
Thalassemia
Thalassemia er mengi arfgengra blóðsjúkdóma sem hafa áhrif á framleiðslu hemóglóbíns (próteinið sem ber ábyrgð á flutningi súrefnis). Þeir eru mismunandi að alvarleika: sumir valda engin einkennum á meðan aðrir eru lífshættulegir. Beinmergsígræðsla er talin í alvarlegustu tilfellunum.
Thalassemia, hvað er það?
Skilgreining á thalassemia
Thalassemia einkennist af galla í framleiðslu á blóðrauða. Til að minna á, er blóðrauði stórt prótein sem er til staðar í rauðum blóðkornum (rauðum blóðkornum) sem hefur það hlutverk að tryggja flutning dixoygen frá öndunarfærum til annarra hluta líkamans.
Sagt er að hálsbólga sé sjúkdómur í blóði. Flutningsvirkni rauðra blóðkorna er skert sem getur haft alvarlegar afleiðingar á líkamann. Á þessum tímapunkti er mikilvægt að hafa í huga að það eru til nokkrar gerðir af thalassemia sem hafa ekki sömu eiginleika eða sama alvarleikastig. Sumir hafa engin einkenni á meðan aðrir eru lífshættulegir.
Orsakir thalassemia
Thalassemia eru erfðasjúkdómar. Þau eru vegna breytinga á einu eða fleiri genum sem taka þátt í myndun blóðrauða, og nánar tiltekið af breytingum á genum sem taka þátt í framleiðslu á blóðrauða próteinkeðjum. Það eru fjórar af þessum: tvær alfa keðjur og tvær beta keðjur.
Hver þessara keðja getur verið fyrir áhrifum í thalassemia. Við getum líka greint:
- alfa-talassemi sem einkennist af breytingu á alfakeðjunni;
- beta-talassemi sem einkennist af breytingu á beta-keðjunni.
Alvarleiki alfa- og beta-þurrkablæðingar fer eftir fjölda gena sem er breytt. Því mikilvægara sem það er, því meiri er alvarleiki.
Greining á thalassemia
Greining á hálsbólgu er gerð með blóðprufu. Heildar blóðtalning gerir það mögulegt að meta útlit og fjölda rauðra blóðkorna og vita þannig heildarmagn blóðrauða. Lífefnafræðilegar greiningar á blóðrauða gera það mögulegt að greina alfa-thalassemia frá beta-thalassemium. Að lokum gera erfðafræðilegar greiningar það mögulegt að meta fjölda breyttra gena og skilgreina þannig alvarleika sjónhimnubólgu.
Einstaklingar sem hafa áhyggjur
Thalassemia er arfgengur erfðasjúkdómur, það er að segja smitast frá foreldrum til barna þeirra. Þeir ná aðallega til fólks frá Miðjarðarhafsbrúninni, Miðausturlöndum, Asíu og Afríku sunnan Sahara.
Í Frakklandi er algengi alfa-thalassemia áætlað 1 af hverjum 350 einstaklingum. Tíðni beta-thalassemia er metin á 000 fæðingar á hverja 1 á ári um allan heim.
Einkenni thalassemia
Einkenni thalassemia eru mjög mismunandi eftir tilfellum og ráðast aðallega af því hversu mikil breyting er á genunum sem taka þátt í framleiðslu á blóðrauða próteinkeðjum. Thalassemia getur verið einkennalaus í minniháttar formum og verið lífshættuleg í alvarlegri mynd.
Einkennin sem nefnd eru hér að neðan varða aðeins millistig til helstu tegunda thalassemiu. Þetta eru bara helstu einkennin. Mjög sértæk einkenni geta stundum komið fram eftir því hvers kyns talassemíur eru.
Blóðleysi
Dæmigert merki um thalassemia er blóðleysi. Þetta er skortur á blóðrauða sem getur leitt til þess að mismunandi einkenni birtast:
- þreyta;
- andstuttur;
- föllitur;
- vanlíðan;
- hjartsláttarónot.
Styrkur þessara einkenna er breytilegur eftir alvarleika thalassemiu.
Gula
Fólk með thalassemia gæti verið með gulu (gulu) sem er sýnilegt á húð eða augnhvítu.
Gallsteinar
Einnig má sjá steinamyndun inni í gallblöðrunni. Útreikningar eru eins og „litlir smásteinar“.
Svæfileiki
Miltastækkun er stækkun milta. Eitt af hlutverkum þessa líffæris er að sía blóðið og sía skaðleg efni þar á meðal óeðlileg rauð blóðkorn. Í thalassemia er milta mjög virkt og stækkar smám saman að stærð. Sársauki gæti fundist.
Önnur, sjaldgæfari einkenni
Sjaldgæfara geta alvarlegar gerðir af thlassemiu leitt til annarra frávika. Til dæmis má sjá:
- lifrarstækkun, það er aukning á stærð lifrarinnar;
- vansköpun í beinum;
- seinkun á þroska barna;
- sár.
Meðhöndlun thalassemia er nauðsynleg til að takmarka tilvik þessara fylgikvilla.
Meðferð við thalassemia
Meðhöndlun á hálsbólgu fer eftir mörgum breytum, þar á meðal tegund thlassemiu, alvarleika hennar og ástandi viðkomandi. Smávægilegustu formin þurfa ekki meðferð á meðan alvarlegu formin krefjast mjög reglubundins lækniseftirlits.
Meðferðin sem nefnd er hér að neðan varða aðeins millistig til helstu tegunda thalassemia
Leiðrétting á blóðleysi
Þegar skortur á blóðrauða er of mikill er nauðsynlegt að gefa reglulega blóðgjöf. Þær felast í því að sprauta viðkomandi blóði eða rauðum blóðkornum frá gjafa til að viðhalda ásættanlegu magni rauðra blóðkorna í blóðinu.
B9 vítamín viðbót
Það gæti verið mælt með því að hefja daglega vítamín B9 viðbót vegna þess að þörfin fyrir þetta vítamín eykst þegar um er að ræða talassemi. B9 vítamín tekur þátt í framleiðslu rauðra blóðkorna.
Ristnám
Miltanám er skurðaðgerð fjarlæging á milta. Þessi aðgerð má íhuga þegar blóðleysið er mjög mikilvægt.
Meðferð við ofhleðslu járns
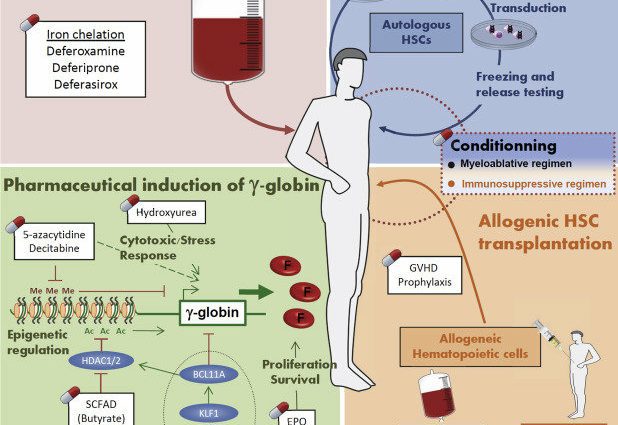
Fólk með thalassemia hefur of mikið af járni í líkamanum. Þessi uppsöfnun getur leitt til mismunandi fylgikvilla. Þetta er ástæðan fyrir því að boðið er upp á járnklóara til að fjarlægja umfram járn.
Beinmergsígræðsla
Beinmergsígræðsla er eina meðferðin sem getur varanlega læknað thalassemia. Þetta er þung meðferð sem aðeins er boðin við alvarlegustu formum sjúkdómsins.
Koma í veg fyrir thalassemia
Thalassemia er arfgengur erfðasjúkdómur. Það er engin fyrirbyggjandi aðgerð.
Á hinn bóginn gera erfðafræðilegar prófanir kleift að greina heilbrigða burðarbera (fólk sem hefur eitt eða fleiri breytt gen(er) en er ekki veikur). Upplýsa skal nokkra heilbrigða burðarbera um hættuna á því að fæða barn með tálasýki. Í sumum tilfellum getur erfðafræðingur metið þessa áhættu. Fæðingargreining getur einnig komið til greina við ákveðnar aðstæður. Það ætti að ræða það við lækninn þinn.