









Efnisyfirlit
Wilsons-sjúkdómur
Hvað er það ?
Wilsonssjúkdómur er arfgengur erfðasjúkdómur sem kemur í veg fyrir brotthvarf kopar úr líkamanum. Uppsöfnun kopar í lifur og heila veldur lifrar- eða taugasjúkdómum. Algengi Wilsons sjúkdóms er mjög lágt, um 1 af hverjum 30 einstaklingum. (000) Það er til áhrifarík meðferð við þessum sjúkdómi, en snemmgreining hans er erfið vegna þess að hann er þögull í langan tíma.
Einkenni
Koparsöfnun hefst við fæðingu en fyrstu einkenni Wilsonssjúkdóms koma oft ekki fram fyrr en á unglings- eða fullorðinsárum. Þau geta verið mjög fjölbreytt vegna þess að uppsöfnun kopar hefur áhrif á nokkur líffæri: hjarta, nýru, augu, blóð... Fyrstu einkenni eru lifrar- eða taugafræðileg í þremur fjórðu tilfellum (40% og 35% í sömu röð) en þau geta verið lifrar- eða taugafræðileg. einnig vera geðræn, nýrna-, blóðmeinafræðileg og innkirtlafræðileg. Lifrin og heilinn eru sérstaklega fyrir áhrifum vegna þess að þau innihalda þegar náttúrulega mestan kopar. (2)
- Lifrarsjúkdómar: gula, skorpulifur, lifrarbilun ...
- Taugasjúkdómar: þunglyndi, hegðunartruflanir, námserfiðleikar, tjáningarerfiðleikar, skjálfti, krampar og samdrættir (dystonia) …
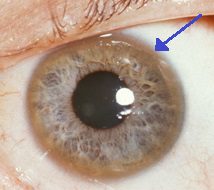
Keyser-Fleisher hringurinn sem umlykur lithimnuna er einkennandi fyrir uppsöfnun kopar í auganu. Auk þessara bráða einkenna getur Wilsons sjúkdómur komið fram með óeinkennandi einkennum eins og almennri þreytu, kviðverkjum, uppköstum og þyngdartapi, blóðleysi og liðverkjum.
Uppruni sjúkdómsins
Við upphaf Wilsons sjúkdóms er stökkbreyting í ATP7B geninu sem staðsett er á litningi 13, sem tekur þátt í efnaskiptum kopars. Það stjórnar framleiðslu á ATPase 2 próteini sem gegnir hlutverki við að flytja kopar frá lifur til annarra hluta líkamans. Kopar er nauðsynleg byggingarefni fyrir marga frumustarfsemi, en umfram kopar verður hann eitraður og skemmir vefi og líffæri.
Áhættuþættir
Smit Wilsons sjúkdóms er sjálfhverf víkjandi. Því er nauðsynlegt að fá tvö eintök af stökkbreytta geninu (frá föður og móður) til að þróa sjúkdóminn. Þetta þýðir að karlar og konur eru jafn útsettar og að tveir foreldrar sem bera stökkbreytta genið en eru ekki veikir eiga á hættu í fjórum við hverja fæðingu að smita sjúkdóminn.
Forvarnir og meðferð
Það er til áhrifarík meðferð til að stöðva framgang sjúkdómsins og draga úr eða jafnvel útrýma einkennum hans. Það er líka nauðsynlegt að það sé hafið snemma, en það tekur oft marga mánuði eftir upphaf einkenna að greina þennan þögla sjúkdóm, lítt þekktan og þar sem einkennin benda til margra annarra sjúkdóma (lifrarbólga sem er lifrarskemmdir og þunglyndi vegna geðræns þáttar) .
„Klóbindandi“ meðferð gerir það mögulegt að laða að kopar og útrýma honum í þvagi og takmarka þannig uppsöfnun hans í líffærunum. Það er byggt á D-penicillamíni eða Trientine, lyfjum sem tekin eru um munn. Þau eru áhrifarík en geta valdið alvarlegum aukaverkunum (nýrnaskemmdum, ofnæmisviðbrögðum osfrv.). Þegar þessar aukaverkanir eru of mikilvægar grípum við til gjafar á sinki sem mun takmarka frásog kopars í þörmum.
Lifrarígræðsla getur verið nauðsynleg þegar lifrin er of skemmd, sem er raunin fyrir 5% fólks með Wilson-sjúkdóm (1).
Systkinum viðkomandi einstaklings er boðið upp á erfðaskimunarpróf. Það gefur tilefni til árangursríkrar fyrirbyggjandi meðferðar ef erfðafræðilegt frávik greinist í ATP7B geninu.